At Charles University, Prague, 180 participants were screened, and 40 were accepted to participate in the study and were examined according to the complex clinical protocol (in this third year we recruited mainly patients with non-AD dementia linked to the planned FTLD paper). In the third year, we analyzed 360 biofluid samples from individuals recruited in the MitAD study. We have continued to measure five mitophagy biomarkers – PINK1 (MyBioSource, MBS7607221), ULK1 (FineTest, EH4191), BNIP3L (FineTest, EH6731), REST (MyBioSource, MBS9354282) and TFEB (MyBioSource, MBS7612687) – these markers together should reflect all five steps of mitophagy process. Additionally, we analyzed standard AD biomarkers – Aβ 1-42, Aβ 1-40, total tau, p-tau 181 (EuroImunn), and a marker of neurodegeneration – neurofilament light chain (UmanDiagnostics) – in another 120 individuals to define the study cohort.
At the University of Oslo, Norway, we have finalized the wet laboratory study investigating roles of ULK1 in neuroprotection. We especially focused on the molecular mechanisms on how ULK1 inhibits AD pathologies: we show that ULK1 increases microglial phagocytosis to eliminate Abeta plaques in the ULK1;5xFAD mice; congruently, ULK1 reduces pTau activities by eliminating kinases that phosphorylate Tau. We have prepared a paper entitled ‘Age-dependent autophagy impairment risks Alzheimer’s disease due to ULK1 reduction’. The entire manuscript was uploaded to the application.
Publications
Oslo team also participated in two original papers 1) In the paper on kynurenic acid in AD, we found that the NAD+ de novo synthetic pathway intermediate kynurenic acid inhibits AD progression (Knapskog AB et al., Alzheimer’s Dement 2023, IF = 14.7) 2) another NAD+ de novo synthetic pathway intermediate quinolinic acid risk death of patients with delirium (Watne LO, et al., J Clin Invest 2023, IF = 15.9).
We published two review papers focused on 1) chemical mitophagy modulators and drug development (Chemical mitophagy modulators: Drug development strategies and novel regulatory mechanisms; Pharmacological Research, IF=9.3 2) the relation between mitophagy and neuroinflammation (Mitophagy and Neuroinflammation: A Compelling Interplay; Current Neuropharmacology, IF = 5.3) and an editorial (Editorial: Mitophagy in health and disease, volume II; Frontiers in Cell and Developmental Biology, IF = 5.5). All papers were published in impacted journals indexed in Web of Science, and the dedication to the Kappa project was acknowledged.
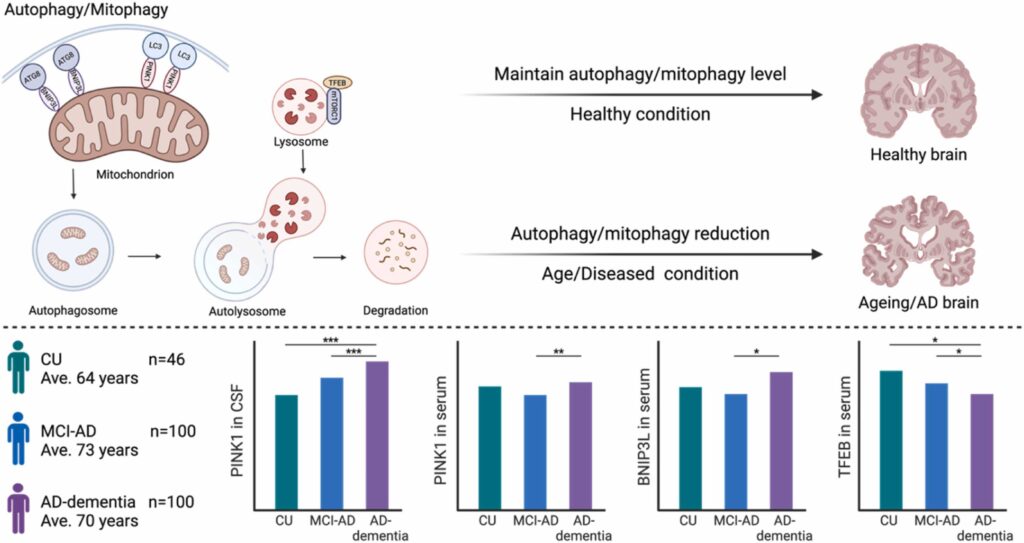
In September, we submitted a research article Alterations of human CSF and serum-based mitophagy biomarkers in the continuum of Alzheimer’s disease in the Autophagy journal (IF=13.3). Currently, the paper is in the second round of revision, with very favorable feedback from the reviewers. In this study, we quantified biomarkers of mitophagy/autophagy and lysosomal degradation (PINK1/BNIP3L and TFEB, respectively) in CSF and serum from 246 individuals, covering mild cognitive impairment due to AD (MCI-AD, n=100), dementia due to AD (AD-dementia, n=100), and cognitively unimpaired individuals (CU, n=46), recruited from the Czech Brain Aging Study. Cognitive function and brain atrophy were also assessed. Our data show that serum and CSF PINK1 and serum BNIP3L were higher, and serum TFEB was lower in individuals with AD than in corresponding CU individuals. Additionally, the magnitude of mitophagy impairment correlated with the severity of clinical indicators in AD patients. Our study reveals mitophagy impairment reflected in biofluid biomarkers of individuals with AD and associated with more advanced AD pathology.
The research article, Serum PAI-1/BDNF Ratio is increased in Alzheimer’s disease and correlates with disease severity, was published in October in ACS Omega Journal (IF= 4.13). In this study, we examined levels of proteins included in neuroprotective molecular pathways (BDNF, PAI-1, tPA) in the serum of patients with dementia due to AD, MCI due to AD, and cognitively unimpaired individuals. We found that BDNF serum levels are lower (13.7% less), and PAI-1 levels are higher in Alzheimer patients with dementia than in Alzheimer patients with amnestic mild cognitive impairment patients (23% more) or controls (36% more). Furthermore, the PAI-1/BDNF ratio was significantly increased in Alzheimer patients as compared to amnestic mild cognitive impairment (36.4% more) and controls (40% more). Lastly, the PAI-1/BDNF ratio negatively correlated with cognition. The study analysed the samples collected in MitAD project and used also the clinical data from the project.
The second research article, Plasminogen activator inhibitor-1 serum levels in frontotemporal lobar degeneration, was accepted by the Journal of Cellular and Molecular Medicine (IF= 5.3) in October. In this research article, we investigated whether PAI-1 and its counter-regulatory tissue plasminogen activator (tPA) are altered in the serum of patients with dementia due to frontotemporal lobar degeneration (FTLD). Thirty-five FTLD patients (21 in the mild cognitive impairment stage (MCI) and 14 in the dementia stage) and 10 cognitively healthy controls were recruited. Serum PAI-1 levels were elevated in the FTLD dementia group as compared to FTLD MCI and controls and also negatively correlated with the cognitive measures. The study analysed the samples collected in MitAD project as well and used also the clinical data from the project.
Both articles were dedicated to the MitAD project and uploaded to the online open repository Zenodo.
(Invited) Review article Diagnostika Alzheimerovy nemoci a ostatních neurodegenerativních onemocnění pomocí biomarkerů z mozkomíšního moku a krve for a Czech journal Neurologie pro praxiwas also accepted in October 2023. In this review paper, we summarized the current overview of biofluid biomarkers in AD. We described the current state of knowledge in diagnosing AD: the golden standard of CSF biomarkers, the biomarkers of non-specific processes that accompany neurodegenerative diseases, and proposed the future use of experimental biomarkers such as mitophagy markers.
Conferences
In July, the MitAD team attended the Alzheimer’s Association International Conference (AAIC) in Amsterdam, Netherlands. Dr. Veverova and Assoc. Prof. Vyhnálek presented a poster Mitophagy biomarkers are changed in Alzheimer’s disease continuum, where she summarized the findings of mitophagy markers in the AD continuum. The results were later on used for the paper Alterations of human CSF and serum-based mitophagy biomarkers in the continuum of Alzheimer’s disease that was submitted recently. MSc. Katonova presented a poster with the title Levels of mitophagy biomarkers differ between individuals with Alzheimer’s disease and frontotemporal lobar degeneration. In this research, we identified different patterns in levels of mitophagy biomarkers in individuals with FTLD compared to AD and controls. Currently, we are preparing the manuscript elaborating on these preliminary findings.
On 18-19 September 2023, the MitAD team co-organized the 1st Norway-UK joint meeting on aging and dementia in Oslo, hosted by Evandro F. Fang (the University of Oslo and Akershus University Hospital, Norway), Lynne Cox (University of Oxford, UK) and Richard Siow (King’s College London, UK). The meeting was opened by educational and political leaders of the two nations, including Prof. Per Morten Sandset (Vice-Rector for Research and Innovation of the University of Oslo), Clare Filshie (Deputy Head of Mission from the British Embassy in Oslo), and Øystein Lund (Counsellor for Research and Education from the Royal Norwegian Embassy in London). Assoc. Prof. Vyhnalek and Dr. Veverova presented their findings from the MitAD project entitled Alterations of human CSF and serum-based mitophagy biomarkers patients from Czech Brain Aging Study (CBAS). The meeting attracted over 40 experts and scholars from 13 countries, including China, Denmark, Greece, Japan, Norway, the United Kingdom, the United States, and others. They gathered to share the latest research findings on various aspects of global ageing and to engage in discussions and workshops on topics including mechanisms of human ageing (such as genetic risk factors, DNA damage and repair, mitophagy and autophagy, etc), connections between ageing processes and disease, and promoting a healthy ageing environment. including. Importantly, discussions included potential interventions to reduce biological ageing and translation of research findings into clinical application. The meeting summary with a title:Meeting summary of The NYO3 5th NO-Age/AD meeting and the 1st Norway-UK joint meeting on ageing and dementia: recent progress on the mechanisms and interventional strategies will be published in the Journal of Gerontological series A (IF = 5.1).
Additionally, several lectures were presented and dedicated to the MitAD project last year.
Prof. Evandro F. Fang visited the Second Faculty of Medicine, Charles University in Prague in June 2023. During his visit, he delivered his lecture on novel mechanisms activating mitophagy from the brain at the Department of Neurology. It was an excellent opportunity for clinicians to learn new approaches to preventing aging and AD. On this occasion, he gave an interview to a hospital magazine, Motol IN, intended for patients and the general public.
In September, Dr. Veverova presented results from the MitAD project during the invited lecture The role of mitophagy in AD pathophysiology at ADDIT-CE: AD-workshop 2 in Brno, Czech Republic. This workshop was attended by 40 scientists.
In another invited lecture, Dr.Veverova, presented the concept of blood-based biomarkers in AD research Krevní biomarkery Alzheimerovy nemoci – budoucnost na dohled? at the 6th Ostrava Liquor Symposium. This symposium is organized for biochemists and clinicians to share the latest developments in liquor diagnosis of various neurological diseases.
An undergraduate student of the MitAD team, Mrs. Alzbeta Katonova, defended her master thesis entitled Mitophagy biomarkers in the continuum of Alzheimer’s disease, supervised by Dr. Veverova and Assoc. Prof. Vyhnalek, with an excellent evaluation. This thesis was chosen out of 244 master theses in the top 10 for the Werner von Siemens Award.