
Alzheimer’s disease, or rather Alzheimer-Fischer disease?
According to Alzheimer’s Disease International, there is a new dementia case every 3 seconds. In 60% of these cases, it is Alzheimer’s disease, a disease typically associated with old age, that causes the dementia syndrome. Unfortunately, as the population ages, we can expect that the number of sufferers will continue to increase in the coming years and that the already unforgiving interval of 3 seconds will gradually shorten. However, Alzheimer’s disease affects not only the patients and their loved ones. Considering the nature of care and its costs, it also has societal implications.
General characteristics
It sneaks into a person’s life, at first completely unnoticed. Pathological processes in the brain take place many years to decades before the first symptoms develop, but only become apparent when they cause significant disruption and death of specific areas in the brain. One of the first to be affected is the hippocampus, a structure resembling a seahorse located in the depth of the temporal lobe, which is responsible for forming and storing recent memories of events and creating a spatial map of the environment. For this reason, the patient first experiences difficulties remembering recent information, recalling plans for the next day, and requires more effort to orientate in an unfamiliar environment. Hand in hand, as the disease persistently spreads in the brain and disrupts the functioning of other brain structures, difficulties with orientation in time and other cognitive (mental) functions follow. In the last stage, the disease affects the entire brain tissue, and the cognitive impairment is so severe and extensive that the patient loses the ability to provide for his or her basic needs independently. The world becomes an incomprehensible and dangerous place for these patients, and they may have difficulties communicating their needs in a comprehensible way. These changes can be one source of feelings of depression, anxiety, and sometimes fearfulness, suspiciousness of the environment, agitation, or aggression. They usually develop in more advanced stages of the disease and are also a direct result of disrupted functioning of the brain areas responsible for mood and behavior. The stage of mild cognitive impairment represents a stage when the first difficulties are already tangible and demonstrable by current diagnostic procedures, yet the person can still function independently in everyday life. When the person becomes fully dependent on the care of loved ones, he/she reaches the stage of dementia syndrome.
Historical background and the development of scientific knowledge
Dementia syndrome is just the tip of an iceberg, preceded by a cascade of pathological events at the level of brain tissue. In 1907, Alois Alzheimer, a German psychiatrist and neuropathologist, first put forward a biological theory of a mysterious mental illness, which was till then thought to have a purely psychological cause. In the brain tissue of his patient Augusta D., who had suffered from dementia before her death, he found the so-called senile plaques and the previously unknown tangles. To the annoyance of the professional community at that time, he linked them to the manifestations of the disease of the now world-famous Augusta D. In the same year, Oskar Fischer, a Czech psychiatrist and neuropathologist, also published the same discovery, but in 16 patients with dementia. However, because of the social circumstances of that time, his name did not become world-renowned.
Since the beginning of the 20th century, a lot of human effort and financial resources have been devoted to trying to spread this knowledge with a clear goal – to find a cure for Alzheimer’s disease. However, to achieve it, it is essential to know the disease in depth, only then we can aim the therapeutic molecules in the right direction. Today, we know that Alzheimer’s disease is almost a lifelong disease, but a person typically notices it at the onset of old age (typically after the age of 65). The first pathological changes at the molecular level take place about 15-20 years before the person or his/her relatives notice any difficulties. Thanks to the development of modern technology, we can also identify these changes in the brain of a patient this early. However, advances in diagnostics have outpaced drug development by leaps and bounds. This is one reason these modern – relatively invasive and expensive – technologies are not yet part of the standard diagnostic process. So what good can such early detection of a disease do? Many promising drugs have failed. But even failure has brought many lessons. Maybe the reason for failure has to do not with the drug itself, but with who received the drug. A retrospective evaluation of some clinical trials found that up to half of the patients enrolled in the study did not suffer from Alzheimer’s disease at all. Perhaps that’s why the drug failed. And perhaps that is why we now have more hope that, thanks to new technologies that allow very early identification of the ‘right’ people, some kind of drug will finally succeed.
Current diagnostic and therapeutic options
What are the current treatment options? There is no drug that can stop the disease completely (a so-called causal treatment). There is currently a so-called symptomatic treatment which aims to slow down the progression of the disease and thus delay the final stage of dementia in which the person loses independence. It helps to ease the specific symptoms and thus reduce the impact of the disease on the quality of life of the patients and their carers. For these reasons, it is best to give the medication to the patient early, when the brain is not yet too damaged by the disease, but notable difficulties are already present. It is worth seeking medical attention when a person notices some difficulties, even more so if someone close to the person notices them and the difficulties worsen over time. The aim of the diagnostic process is to distinguish between difficulties that are a normal part of the natural aging process and those that are more pronounced and therefore probably a result of a disease. We however cannot resolve this during a single visit to the doctor. At the very least, a detailed analysis of the patient’s difficulties for which he or she is coming, a thorough neurological examination, brain imaging and a neuropsychological examination which usually takes about two hours, are important.
Prevention options – in old age and overall
Non-pharmacological therapeutic approaches also play a key role in the prevention, onset, and over the course of Alzheimer’s disease. Factors such as physical activity, diet or meditation can often do more than we would expect. Many studies have found that regular aerobic activity (i.e. activity during which our heart rate is approximately 110-120 beats per minute) can substantially reduce the risk of developing Alzheimer’s disease. Older people without physical activity had twice the risk of developing dementia compared to those with regular physical activity. More active individuals also appear to have better memory and are slower to experience brain tissue loss. Exercise increases blood flow to the brain and protects blood vessels from storing fat in their walls. This protects the brain and its functions from damage. Another important factor for brain health is a balanced and vitamin-rich diet. Such a diet should include a regular supply of fresh fruit and vegetables, food rich in fatty acids (natural oils, nuts, sprouts, fresh fish), a sufficient intake of legumes and medicinal spices (e.g. turmeric). On the other hand, we should avoid simple sugars, preservatives, artificial sweeteners and flavorings, solid fats, and excessive alcohol in our diet. An ideal example of a diet is the so-called Mediterranean diet. Finally, we should not forget to mention mental health care, whether in the form of meditation or spirituality. Cultivating and maintaining good social relationships with friends and family is also important for mental health. Long-term loneliness in old age increases the risk of heart disease, stroke, and is associated with a higher incidence of Alzheimer’s disease.
Summary information window – important numbers and terms:
- Alzheimer’s disease, alternatively Alzheimer-Fischer disease;
- 1907: the year in which the biological basis of a formerly mysterious mental illness was first documented by Alois Alzheimer and, independently of him, by Oskar Fischer;
- 60%: the percentage of cases of dementia syndrome for which Alzheimer’s disease is responsible;
- mild cognitive impairment: a stage of the disease in which a person already has noticeable cognitive difficulties (e.g. with memory) but can function independently in everyday life. It is sometimes referred to as an intermediate stage between healthy aging and dementia;
- dementia: a set of symptoms including cognitive impairment, behavioral changes and loss of independence in activities of daily living; always a manifestation of brain disease;
- senile dementia: a term that is a relic of an earlier period and should no longer be used today; in the current state of knowledge, we now know that dementia is not an inevitable manifestation of aging, but always a manifestation of a disease;
- neurodegenerative disease: a disease causing the gradual death (-degeneration) of brain tissue (neuro-);
- hippocampus: a brain structure crucial for the formation of new memories and the connections between them; its function is the first to be affected by Alzheimer’s disease;
- beta amyloid and tau: proteins; the deposition of their pathological forms in brain tissue is – according to current knowledge – at the beginning of Alzheimer’s disease, many years before the development of the first symptoms.
References
- Alzheimer’s Disease International. (2019). World Alzheimer Report. London.
- Alzheimer’s Disease International. (2018). World Alzheimer Report 2018. London.
- Čechová, K., Fendrych Mazancová A., Marková H. (Eds.) et al. (2019). V bludišti jménem Alzheimer: Na co v ordinaci nezbývá čas. Prague: Albatros Media.
Mitophagy in neurodegeneration and aging
Invited article for BestPractice
Ruben Gudmundsrud
Molecular biology and biochemistry programme, University of Oslo
Hilde Nilsen
Department of Clinical Molecular Biology, University of Oslo and Akershus University Hospital, 1478 Lørenskog, Norway
Vilhelm A. Bohr
Laboratory of Molecular Gerontology, National Institute on Aging, National Institutes of Health, Baltimore, MD 21224
Danish Center for Healthy Aging, University of Copenhagen, Blegdamsvej 3B, 2200 Copenhagen, Denmark
Evandro F. Fang
Department of Clinical Molecular Biology, University of Oslo and Akershus University Hospital, 1478 Lørenskog, Norway
Corresponding author
Evandro F. Fang (e.f.fang@medisin.uio.no)
Introduction: the formidable pressure of aging and age-related neurodegeneration
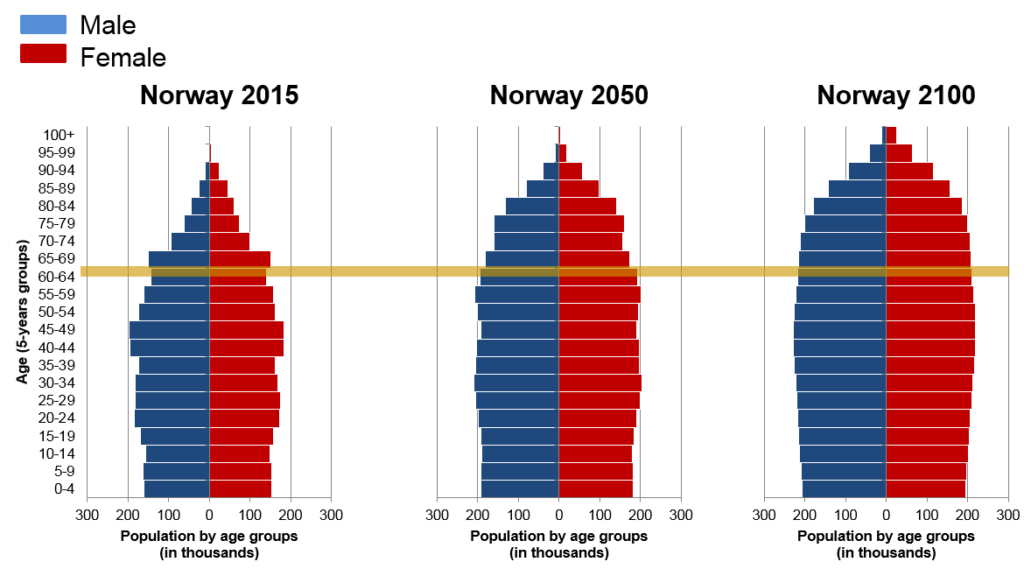
People worldwide are living longer. It gives a pressure of formidable healthcare challenges brought about by the challenges of aging. In Norway, there was 16.6% of elderly (65+) in 2016, and this will increase dramatically by 2050 and 2100 (Figure 1). Aging is the primary driver of almost all the non-genetic human diseases, including neurodegeneration. As the most common form of dementia, Alzheimer’s disease (AD) affects 44.5 million people in the world, which is expected to be tripled by 20501. This situation poses large socioeconomic problems, including expenditure on geriatric care, heavy load for the family and the devastating loss of life quality for affected individuals1. There is no cure for AD. Further understanding of its sophisticated molecular mechanisms will propel novel drug discovery, with broad economic impact.
How mitophagy is defective, and how it links to aging and neurodegeneration
Mitochondria are the “powerhouses of the cell”. They play fundamental roles in life and health, including energy metabolism, major cell signaling pathways, cross-talks between mitochondria and nucleus, neuronal development and even cell fate determination (death or survival)2. Thus, human health can be directly affected by the mitochondrial quality that is tightly regulated by several cellular pathways, majorly the mitochondrial autophagy pathway.
Autophagy is a cellular process to degrade and further recycle broken organelles or macromolecules. There are different forms of autophagy, for example macroautophagy which is non-specific and commonly used for production of energy during starvation or exercise. Mitophagy is degradation of mitochondria, in other words: Selective autophagy of mitochondria1, 3. The process of autophagy can be outlined roughly as follows: Encapsulation (nucleation) of material to be degraded in a double membrane enclosed organelle into a autophagosome (mitophagosome in mitophagy), which is then degraded in a fusion process with the lysosome to create the autolysosome (or mitolysosome in mitophagy)1, 2. Mitophagy is often initiated by damaged mitochondria which pose a threat to cells. Damaged mitochondria leak ions, electrons and produce less energy. Electrons form reactive oxygen species (ROS), which cause oxidative stress. Accumulation of dysfunctional mitochondria is implicated in neurodegeneration1, 2, 4.
Defective mitophagy in AD may contribute to accumulation of damaged mitochondria, which further interacts with the formation of Aβ plaques and Tau tangles (two hallmarks of AD), and finally leading to AD progression. First, mitochondrial dysfunction is a common phenomenon is both sporadic and familiar AD5. Second, defective mitophagy has been shown in AD iPSC-derived neurons and AD mice1. Third, accumulation of damaged mitochondria can induce formation of Aβ plaques and Tau tangles, and vice versa1. E.g., defect mitophagy stress the cell which accelerates the formation of Ab plaques, this might be the result of the mitochondrial oxidative stress, which increase the production of amyloid b (Ab) by the modification of g-secretase (enzyme complex which cleaves part of APP to make Ab)6. Mitochondria in AD affected cells seems to obtain damage and aberrant mitophagy processing. This points to a vicious cycle, where damaged mitochondria cause AD pathology which again damages more mitochondria1.
During a lifetime, our DNA accumulates damage. This damage activates DNA repair systems. One of the involved proteins is poly(ADP-ribose) polymerase 1 (PARP1), which is involved in several DNA repair pathways. This machinery uses the cofactor nicotinamide adenine dinucleotide (NAD+), which is also the case for part of one of the many mitophagy inducing pathways2. Proteins of the sirtuin (SIRT) family are NAD+-dependent deacetylases involved in a range of processes, probably also the activation of the mitophagy machinery via signal pathways2. When DNA damage increase (aging, accelerated aging diseases), DNA repair systems (PARP1 in particular) use a considerable part of the NAD+ pool, which again decreases the SIRT1 activity and we observe less mitophagy2. This NAD+ depletion seems to be universal in aging1.
Interventional strategies to upregulate mitophagy
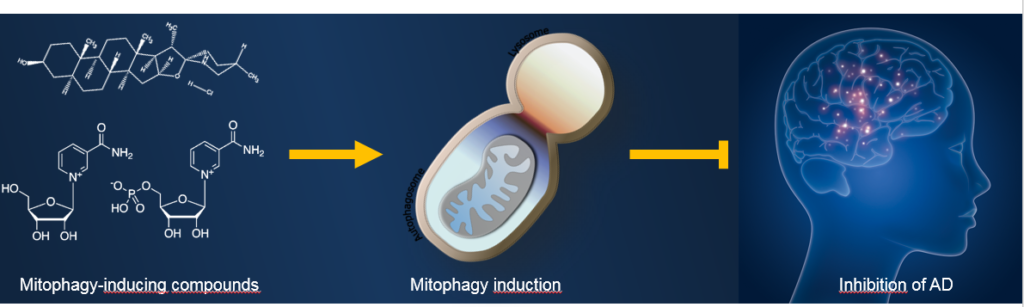
Upregulation of mitophagy to maintain a healthy mitochondrial pool is emerging as a novel therapeutic strategy for AD (Figure 2). E.g., upregulation of autophagy (including mitophagy) through the mammalian target of rapamycin (mTOR) inhibitor rapamycin alleviated cognitive deficits in mice and decreased Ab plaque7. Feeding the 3xTg AD mice with an NAD+ precursor, nicotinamide riboside (NR), alleviated Tau pathology8. Genetic upregulation mitophagy was also able to inhibit AD pathology in mice9. In addition, it would be interesting to test the effect of other known mitophagy inducers, such as tomatidine (a small natural compound from green tomato), in AD mice.
Outstanding questions and clinical translation
For a translational perspective, the authors are now moving forward from bench-top to clinical trials for the NAD+ precursors, on AD, and other age-related diseases, including premature aging diseases with neurodegeneration. There are a couple of clinical trials for NAD+ precursors running. Finding out the right dosage of NAD+ precursors and any side effects are part of the goals for those trials1. Questions remain to be elucidated, like how DNA damage affects mitophagy and why mitophagy is impaired in AD. Do NR and another NAD+ precursor, nicotinamide mononucleotide (NMN) have the same clinical effects?
Conclusion
Ageing and neurodegeneration together pose socioeconomic challenges. New research implicates dysfunctional mitochondria in both ageing and neurodegeneration and the clean-up process of mitophagy seems to be aberrant. Novel substances have shown great potential for possible interventions that could prevent or slow down these processes by inducing mitophagy. Several clinical trials are running to test whether the NAD+ precursor NR could become an effective prophylactic supplement. Tomatidine has also shown great potential for a possible supplement. If these processes are possible to slow down, or even abolish AD and other neurodegenerative diseases, we shall reach to a healthy aging society.
References
1. Kerr, J.S. et al. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci 40, 151-166 (2017).
2. Fang, E.F. et al. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 17, 308-321 (2016).
3. Palikaras, K., Lionaki, E. & Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525-528 (2015).
4. Fivenson, E.M. et al. Mitophagy in neurodegeneration and aging. Neurochem Int 109, 202-209 (2017).
5. Swerdlow, R.H., Burns, J.M. & Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta 1842, 1219-1231 (2014).
6. Gwon, A.R. et al. Oxidative lipid modification of nicastrin enhances amyloidogenic γ‐secretase activity in Alzheimer’s disease. Aging Cell 11, 559-568 (2012).
7. Spilman, P. et al. Inhibition of mTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-β Levels in a Mouse Model of Alzheimer’s Disease. PLOS ONE 5, e9979 (2010).
8. Hou, Y. et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A 115, E1876-E1885 (2018). 9. Du, F. et al. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 140, 3233-3251 (2017).