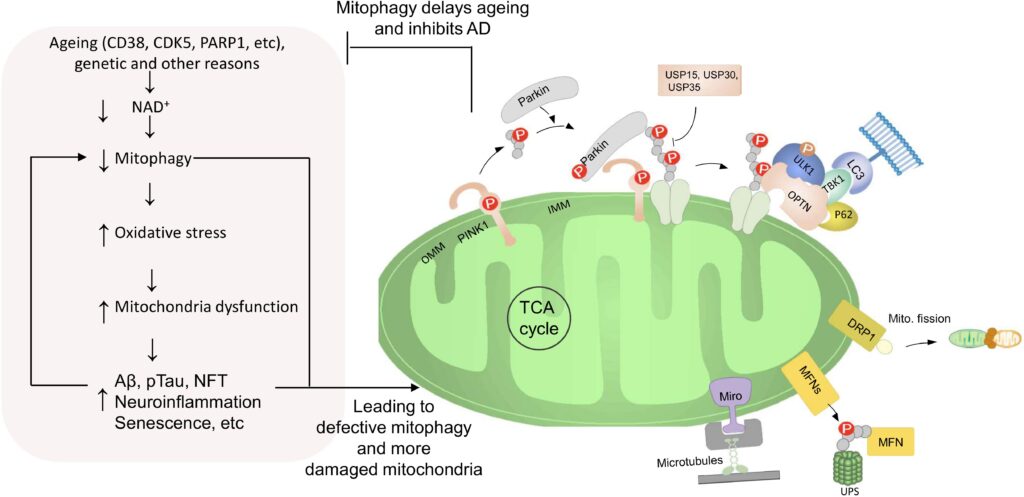
AD is the most prevalent neurodegenerative disease worldwide. The pathological hallmarks are Aβ and phosphorylated tau protein, however, their detection is either invasive or expensive for health care system. Despite extensive research, the exact cause of AD is not fully understood and many drug trials targeting tau and Aβ proteins failed. Mitochondria play a critical role in neuroplasticity, neuronal survival and its dysfunction is a common feature in AD.
Mitochondrial dysfunction occurs prior to development of Aβ plaques and tau tangles and thus might be an early trigger of AD pathology. Conversely, overproduction of Aβ can lead to localization of Aβ to mitochondria and induce dysfunction. Moreover, tau pathology induces mitochondrial dysfunction, impairs mitochondrial axonal transport and further exacerbates Aβ-induced mitochondrial damage in AD. These findings highlight mitochondrial maintenance and the clearance of damaged mitochondria as a necessity for the maintenance of neuronal function. This process is regulated by mitochondrial autophagy, termed mitophagy. Mitophagy specifically eliminate damaged mitochondria or clear all mitochondria during developmental stages or starvation. Mitophagy in neurons is necessary to prevent neuronal death and pathogenic brain aging. Several mitophagy pathways are known, and many are conserved from C. elegans to humans. In addition, a link between autophagy dysfunction and AD has been proposed, but little is known about the relation between mitophagy and AD. Our studies suggest down-regulation of mitophagy contributes to aging and neurodegeneration observed in models of premature aging disease. Recently, we proposed mitophagy as a key driver in AD initiation and progression but specific mitophagy biomarkers in AD need to be determined. Mitophagy markers are promising candidates for early and noninvasive identification of patients at risk of AD.